The norovirus typingtool facilitates early recognition of globally emerging strains or indications of common sources. Regular updating of the reference sequence set and a standardized nomenclature enable timely comparison of sequences found in different laboratoria.
- To the sequence typing tool
- Introduction
- Genotyping process in three steps
- Typing Regions
- Reference set update
Introduction
By issuing preliminary names for new strains and using a
standardized nomenclature, findings in different parts of the world
can be compared.
With this tool sequences of any part of the Norovirus (NoV) genome
can be assigned to a NoV genogroup, and sequences of all commonly
used regions for typing can be assigned to a genotype.
For one specific NoV genotype (II.4) the variant of that
genotype is determined.
Sequences of other Caliciviridae are also recognized by the tool
and are assigned to their (preliminary) genus. In case of
Sapoviruses a genogroup is also assigned.
A paper describing the norovirus typingtool and the enterovirus
typingtool, which was developed using the same platform:
Kroneman A, Vennema H, Deforche K, Avoort HV, Peňaranda S, Oberste
MS, Vinjé J, Koopmans M.
An
automated genotyping tool for enteroviruses and noroviruses. J
Clin Virol. 2011 Jun;51(2):121-5.
Genotyping process in three steps
Step 1:
This initial step performs a nucelotide blast scan, using Blast.
The submitted sequence is blasted against a set of whole genome
sequences for determination of the genus within the Caliciviridae
family and the genogroup (for NoV and for SaV). The start and end
positions of the fragment are determined using whole genome
sequences.
Step 2:
This second step is performed for sequences which were identified as NoV Genogroups I and II in step 1.
ORF1 sequences and ORF2 sequences are analysed seperately using a different reference set. For each submitted sequence that overlaps sufficiently (= 100nt) with either the ORF1 region or the ORF2 region supported by the tool, an alignment is performed using ClustalW and a phylogenetic tree (NJ) is constructed with the HKY851 method using PAUP* software.
HKY85 is a likelyhood method with 100 bootstrap replicates. The alignment contains the sequence together with the pre-aligned reference sequences for each genotype. In the PAUP* log file the bootstrap tree is also presented.
For each genotype two, in some cases more, reference sequences
have been selected, representing the level of variance within the
cluster. If available (and covering the complete regions) the known
prototype strains are used.
Step 3:
When a genotype II.4 is identified in step 2, a second analysis is done to identify a specific variant.
The same phylogenetic method is used as in step 2. Again two representative sequences per subcluster are used in the analysis.
Typing regions
The submitted sequences are analysed separately for ORF1 and
ORF2 type.
The lengths, and positions related to NC_001959 for
Genogroup I (GI) sequences and U07611 for
Genogroup II (GII) sequences of the supported typing regions for
step 2 and 3 are:
Starting position End
position
ORF1 GI
4596
5357
ORF2 GI
5358
7000
ORF1 GII
4323
5084
ORF2 GII
5091
6727
The length of the individual strains may vary due to gaps in the aligned sequence.
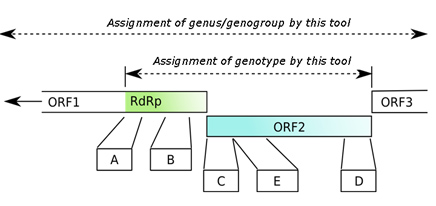
Figure 1: A representation of the complete norovirus genome with an indication of the regions covered by the different analysis steps in the typing tool.
Reference sets update
Information on the latest reference sets, used by the tool, can be obtained at noronet@rivm.nl
Latest updates:
- December 2012: Addition of GII.4 Sydney 2012 variant sequences to the reference set
- December 2012: Revision of the GII.4 variant names according to a new international consensus nomenclature (in press, Archives of Virology, 2013)
- April 2013: Addition of VP1 genotype GI.9 and revision of the ORF1 nomenclature according to consensus nomenclature paper
Conversion of GII.4 variant names
|
Old name |
New consensus nomenclature |
|---|---|
|
Bristol |
Bristol 1993 (non-epidemic cluster) |
|
Camberwell |
Camberwell 1994 (non-epidemic cluster) |
|
1996 |
US 95_96 |
|
2001 |
Kaiso 2003 (non-epidemic cluster) |
|
2002 |
Farmington Hills 2002 |
|
2002CN |
Lanzou 2002 (non-epidemic cluster) |
|
2003 1 |
Asia 20031 |
|
2004 |
Hunter 2004 |
|
2006a |
Yerseke 2006a |
|
2006b |
Den Haag 2006b |
|
2007 |
Osaka 2007 |
|
2008 |
Apeldoorn 2007 (non-epidemic cluster) |
|
2010 |
New Orleans 2009 |
|
NA |
Sydney 2012 2 |
1: variant 2003/Asia 2003 is a recombinant with a GII.4 ORF2 and
a GII.P12 ORF1
2: Variant Sydney 2012 is a recombinant with a GII.4 ORF2 and a
GII.Pe ORF1
Conversion of ORF1 genotype names
|
Old name |
New consensus
|
Comment |
|---|---|---|
|
All |
P added |
In all ORF1 genotypes a 'P' has been added before the character indicating the genotype: GII.4 ->GII.P4 ('P' for 'polymerase') |
|
GI.e |
GI.P9 |
This is the ORF1 genotype of the new GI.9 ORF2 genotype |
|
GII.b |
GII.P21 |
The ORF1 region of new VP1 genotype GII.21 clusters with the earlier defined ORF1 orphan genotype GII.b |
|
GII.d |
GII.P22 |
The ORF1 region of new VP1 genotype GII.22 clusters with the earlier defined ORF1 orphan genotype GII.d |
|
GII.17 |
GII.P13 |
VP1 Genotypes GII.P13 and GII.17 have the same ORF1 genotype: GII.13 |
|
GII.19 |
GII.P11 |
Porcine VP1 Genotypes GII.11 and GII.19 have the same ORF1 genotype: GII.P11 |