You can download the poster presentation 'Geotagging Hepatitis A virus, a dynamic tool for public health surveillance' (PDF) from the download-box.
Text version of the poster
Authors
Rita de Sousa 1,2 , Harry Vennema 1 , Linda Verhoef 1 , Annelies Kroneman 1 , Mariska Pertignani 3 , Jussi Sane 1,4 , Marion Koopmans 1,5 , on behalf of the HAV network.
- National Intstitute for Public Health and the Environment (RIVM), Bilthoven, the Netherlands
- The European porgogramme for Public Health Microbiology training (EUPHEM), European Centre for Disease Prevention and Control (ECDC), Stockholm, Sweden
- Municipal Health Service, Rotterdam, the Netherlands
- European Programme for intervention Epidemiology Training (EPIET), European Centre for Disease Prevention and Control (ECDC), Stockholm, Sweden
- Erasmus Medical Centre, Rotterdam, the Netherlands
Background
- Hepatitis A is an acute liver disease caused by hepatitis A virus (HAV)
- Transmitted by oral-fecal route
- Distributed worldwide, but infection depends on level of country endemicity (related socio-economic conditions and sanitation)
- Three genotypes (I, II, III) divided into A and B subtypes are known two infect humans
- There is a genetic relatedness of HAV with geographical origin
- Hepatitis A cases are mainly seen in travelers to endemic areas, and occasional infection linked to consumption of contaminated imported foods
- The HAV NET database coordinated by RIVM, has been used to help to identify the source or track food-related clusters and outbreaks, improving the capacity of laboratory response, by using molecular and epidemiological information on HAV from different geographic origins.
Aim
Our aim in this study was to analyse the suitability of the currently available hepatitis A database for detection of diffuse international foodborne outbreaks
- Analyse genotyping target regions and length of the sequences available in the data-base.
- Evaluate the proportion of strain sequences that are linked to
a specific country of infection (coverage and representativeness),
HAV genotype diversity in each region, level of
endemicity, mode of infection (e.g travel related) - Create a HAV NET website to diffuse the information about the aims of network and participation.
Methods
Data
Hepatitis A data-base includes HAV sequences and epidemiological information from HAV NET Members combined with additional sequences retrieve from GenBank.
Data Analysis
- Sequence analysis. Analysis of the sequence targets and lengths from HAV NET
- Analysis of combined data (epidemiological and molecular data).
Information linked with sequences was exported to Excel and data was analysed by geographic distribution, country of infection, possible transmission routes, and genotype diversity by geographic region.
Fields in HAV NET database
- Identification of the sequence data: Provider (HAV NET, GenBank)
- Epidemiological data related with human case:
Case identification and immune status.
Possible transmission route (e.g travel related) - Molecular information:
Typed region and length of the fragment - Geographic information of the sequence:
Suspected country of infection
Level of endemicity
Results
Database:
- Total of sequences analysed in the database (N=7120)
- Sequences from HAV NET (N=1634) + Genbank (N=5486)
- Period of time of sampling: 1957-2013
Step 1: Sequence analysis
- Homogeneity in the target region used for typing from sequences from HAV NET members (73% sequences in V1/2A region)
- Different lengths of the fragments (>150 lengths; range 120 nt to 901 nt)

Step 2: Analysis of combined data
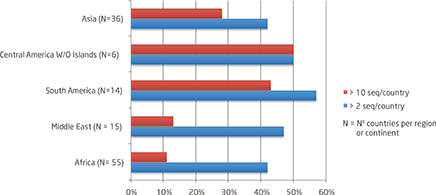
- Considering at least 2 HAV sequences by country, coverage ranges from 42-57% by continent or regions (fig 2)
- Of the total 67% (HAV NET = 942; GenBank =3795) sequences have information on origin of infection (fig 3)
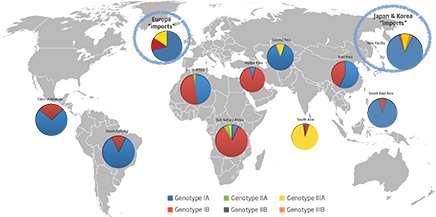
- Genotype IA (60%) is the most prevalent;
- Genotype IB (19.5%) is more prevalent in Africa and Middle East;
- Genotype IIIA (19.7 %) is most prevalent in Asia (India, Afghanistan)
- Information on transmission route was available for 16% of these sequences (table 1)
- Travel related information indicates that the higher number of sequences are from High to High-medium endemic countries; Genotype IB counts for 55% of the sequences, mainly from travellers to North Africa and Turkey.
|
Possible transmission route |
HAV NET (N=637; 67%) |
Genbank (N=104: 2,7%) |
|---|---|---|
|
Travel related |
277 |
51 |
|
Person-to-person (non-sexual contact) |
219 |
3 |
|
Person-to-person (sexual contact) |
102 |
19 |
|
IV drug users |
14 |
11 |
|
Food and waterborne source |
25 |
20 |
Step 3: HAV NET website
Conclusion
- Available sequences have made it possible to identify and linked outbreaks and their geographic origin internationally
- Comparative analyses of sequences data, internationally, is hampered by lack of target standardisation
- The currently available HAV NET database contains information from most continents and genotypes, but resolution in some geographic areas is limited by the number and the length of sequencies.
- The lack of some epidemiological information was evident and can limited for the understanding of HAV infection (e.g.transmission routes of some strains)
Recommendations
- The resolution of HAV genotype-base cluster distribution can be greatly improved by more systematic collection of data, and target standardisation (region and length), through harmonization of genotyping protocols between laboratories.
- Geographic representation and coverage need to be increased to
improve applicability
of the database for support of public health investigations
References
- Jacobsen H. The global prevalence of hepatitis A virus infection and susceptibility: A systematic Review. Geneva: World Health Organization; 2009. Available from: http//whqlibdoc.who.int/hq20107WHO_IVB_10.01_eng.pdf.
- Koopmans et al. Early Identification of common-source foodborne virus outbreaks in Europe. Emerg. Infect. Dis. 2003. 9:1136 – 1142. / Robertson BH et al., Genetic relateness of hepatitis A strains recovered from different geographical regions. J.Gen.Virol. 1992. 73: 1365 -1377