
Auteurs
Nynke Rots1 en Cécile van Els1,2
- RIVM, Centrum Infectieziektebestrijding
- Universiteit Utrecht, Faculteit Diergeneeskunde, Infectieziekten & Immunologie, Departement voor Biomolecular Health Sciences
Infectieziekten Bulletin, december 2023
De belangrijkste lessen van het ontwikkelen van COVID-19-vaccins
Toen het nieuwe, van dier naar mens overgesprongen, erg besmettelijke en ziekmakende coronavirus (severe acute respiratory syndrome)-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2) wereldwijd de respiratoire infectieziekte COVID-19 (COronaVIrus Disease 2019) veroorzaakte, was de behoefte aan een goed vaccin snel duidelijk. De beelden van overvolle ziekenhuizen in Noord-Italië bepaalden het primaire doel van vaccinatie: beschermen tegen COVID-19 gerelateerde ziekenhuisopname.
Voor het ontwikkelen van een vaccin tegen een onbekend virus is het ontrafelen van de genetische code van het virus noodzakelijk. Dankzij nieuwe, snelle technieken werd de code van (severe acute respiratory syndrome)-CoV-2 binnen enkele weken na ontdekking en isolatie van het virus op 11 januari 2020 gepubliceerd. Uit deze code konden de eiwitsamenstelling en structurele eigenschappen van het virus worden afgeleid. Op basis van deze kennis en de kennis over beschermende immuniteit tegen andere coronavirussen zoals SARS-CoV en (Middle East Respiratory Syndrome) (Middle East Respiratory Syndrome virus) konden wetenschappers het belangrijkste doelwit-eiwit voor vaccins snel aanwijzen. Dit was het kenmerkende uitsteeksel op de virusmantel, waarmee het virus een gastheercel kan binnendringen en een infectie kan veroorzaken: het SARS-CoV-2 spike-eiwit.
Wereldwijd werd gestart met het ontwikkelen van COVID-19-vaccins, de meeste gebaseerd op dit spike-eiwit. Vaccinfabrikanten werden hierbij op allerlei manieren, wetenschappelijk en vooral financieel, ondersteund door overheden, universiteiten, internationale gezondheidsorganisaties waaronder de (World Health Organization) (World Health Organisation), en fondsen zoals de Gates Foundation en CEPI (Coalition for Epidemic Prepardness Innovations). CEPI is een publiek-privaat-filantropisch partnerschap dat vaccinontwikkeling financieel ondersteunt en eerlijke toegang tot vaccins bevordert. De WHO had als gezaghebbende en wereldwijd opererende organisatie een belangrijke coördinerende rol in de vaccinontwikkeling. Talloze commissies van experts met kennis over de infectieziektebestrijding en vaccins werden ingesteld. Zij verzamelden kennis en kunde over het nieuwe coronavirus en de ziekte COVID-19 in de verschillende landen en deelden inzichten over kandidaat-vaccins via wetenschappelijke vergaderingen en webinars. Er kwamen richtlijnen voor vaccinontwikkelaars. Zo werd de zogenoemde (Research and Development) Blueprint Novel Coronavirus opgesteld, met daarin een internationaal Target Product Profile (TPP) voor COVID-19-vaccins. Dit is een lijst met productkenmerken van kwaliteit, veiligheid en werkzaamheid waaraan de vaccins moesten voldoen. Ook werd een protocol voor gerandomiseerde gecontroleerde fase III-studies opgesteld, waarmee bescherming tegen COVID-19 kon worden aangetoond. Hiermee konden onderzoeksteams en vaccinfabrikanten aan de slag.
COVID-19 werd op 11 maart 2020 door de WHO uitgeroepen tot pandemie, een wereldwijde epidemie. Later die maand waren er al zo’n 54 kandidaat-vaccins in ontwikkeling, waarvan de eerste 2 zelfs al getest werden in de mens. Tijdens het eerste jaar van de pandemie nam het aantal kandidaat-vaccins in rap tempo toe tot 163 kandidaat-vaccins in preklinische ontwikkeling, waarvan er 51 in de mens getest werden. Op 21 december 2020 kreeg Pfizer/BioNTech als eerste een positieve boordeling van het Europese Medicijn Agentschap (EMA) en kreeg toestemming van de Europese Commissie (EC) voor toelating tot de markt van de Europese Unie (EU) voor hun op mRNA gebaseerde COVID-19-vaccin Comirnaty. Zestien dagen later volgde Spikevax, een tweede mRNA-vaccin van vaccinfabrikant Moderna. Hierna volgden 2 virale vectorvaccins. Op 29 januari 2021 werd Vaxzevria toegelaten tot de Europese markt (van vaccinfabrikant/universiteit combinatie AstraZeneca/Oxford Universiteit) en op 11 maart 2021 Jcovden, ontwikkeld door vaccinfabrikant Janssen (Johnson & Johnson). Het ontwikkelen van op eiwit gebaseerde vaccins kostte iets meer tijd (Box 1).
Hoe werken de in de (Europese Unie) toegelaten COVID-19 vaccins en hoe goed beschermen ze?
De innovatieve vaccinplatforms voor de eerste mRNA-vaccins Comirnaty en Spikevax en de virale vectorvaccins Vaxzevira en Jcovden maken gebruik van de genetische code voor het (severe acute respiratory syndrome)-CoV-2 spike-eiwit. Om deze code in menselijke cellen af te leveren maken mRNA-vaccins gebruik van kleine vetbolletjes als transportmiddel, terwijl de virale vectorvaccins gebruikmaken van een onschadelijk gemaakt verkoudheidsvirus om cellen te infecteren. Na vaccinatie lezen cellen van het lichaam de genetische code en gaan het spike-eiwit maken. De later toegelaten, COVID-19-vaccins Nuvaxovid (fabrikant Novavax, toelating op 20 dec 2021), COVID-19 Vaccine Valneva (fabrikant Valneva, toelating op 24 juni 2022), VidPrevtyn Beta (fabrikant Sanofi, toelating op 10 november 2022), Bimervax (fabrikant HIPRA, toelating op 30 maart 2023) bevatten (o.a.) het spike-eiwit zelf, aangevuld met een hulpstof om de afweerrespons van het lichaam te versterken. Het immuunsysteem herkent bij alle vaccinplatforms het spike-eiwit als lichaamsvreemd, waardoor het hiertegen antistoffen en (geheugen) T-cellen gaat maken. Het vaccin wordt na een tijdje weer afgebroken en opgeruimd. Na een infectie met het coronavirus herkent het immuunsysteem het virus al, waardoor het sneller weggewerkt kan worden en verdere vermenigvuldiging, verspreiding en ziekte voorkomt. Resultaten van de fase III-studies van de eerste vier COVID-19-vaccins lieten in volwassenen een acceptabel veiligheidsprofiel zien en een onverwacht hoge werkzaamheid (efficacy) tegen ziekte, veroorzaakt door de toenmalig circulerende eerste SARS-CoV-2 stammen: 95% (Comirnaty), 94,1% (Spikevax), 74% (Vaxevria), 66% (Jcovden) en 89,3% (Nuvaxovid). Voor de later toegelaten vaccins konden geen fase III-efficacy studies gedaan worden, aangezien vergelijking met een niet gevaccineerde controle groep niet meer mogelijk was, maar werd de werkzaamheid aangetoond door zogenaamde ‘non-inferiority’ immunogeniciteitsanalyses ten opzichte van reeds toegelaten ‘comparator’-vaccins. De echte veiligheidsprofielen van de vaccins werden pas duidelijk na wereldwijde grootschalige inzet in alle leeftijdsgroepen (fase (Informatievoorziening)). Zo bleek de zeer zeldzame bijwerking trombose in combinatie met een tekort aan bloedplaatjes vaker voor te komen bij oudere volwassenen na vaccinatie met een op adenovirus gebaseerd vectorvaccin. Voor myocarditis en pericarditis was dit het geval na mRNA-vaccinatie vooral bij jongvolwassen mannen.
Door voortschrijdende moleculaire evolutie van het SARS-CoV-2-virus, met veranderde besmettelijkheid en ontsnapping aan spike-gerichte immuniteit tot gevolg, worden oorspronkelijke COVID-19-vaccins inmiddels vervangen door aan spike-variant aangepaste vaccins.
Deze ongelooflijke prestatie riep veel vragen op: ‘Kan een vaccin dat zo snel ontwikkeld is veilig zijn?, ‘Is het voldoende getest?’. Normaal gesproken duurt het ontwikkelen van een nieuw vaccin, van ontwerpfase tot markttoelating, 12-15 jaar. Nu lukte het in 11 maanden na het bekendmaken van het virale genoom. Hoe kon dit?
Een aantal unieke factoren en aanpassingen aan alle fasen van het reguliere vaccinontwikkeltraject hebben tot deze uitzonderlijke tijdwinst geleid.
Allereerst hebben wetenschappers, vaccinfabrikanten en medewerkers betrokken bij de infectieziektebestrijding alles uit hun handen laten vallen en zijn collectief aan de slag gegaan met het bestrijden van de pandemie. Daarbij is het aantal mensen dat hierbij betrokken was fors uitgebreid. Vanaf het begin is gegenereerde kennis zo snel mogelijk gedeeld, zoals op preprintservers en tijdens wetenschappelijke (online) bijeenkomsten van experts. Dit betrof de genetische code van het virus, maar ook protocollen van klinische studies en data van (pre)klinische studies.
Ten tweede gaf de (World Health Organization) snel richtlijnen voor het eindproduct en de testprocessen, die centrale sturing en helderheid gaven aan alle vaccinontwikkelaars.
Ten derde kon wetenschappelijke kennis en kunde uit eerdere ontwikkeltrajecten gebruikt worden. Zo was men al ver met het ontwikkelen van innovatieve, snel te produceren mRNA-vaccins en virale vectorvaccins, zoals tegen ebola of ziekte veroorzaakt door (severe acute respiratory syndrome)-CoV-2 verwante virussen zoals SARS-CoV en (Middle East Respiratory Syndrome-coronavirus). Hierdoor was snel bekend dat het SARS-CoV-2 spike-eiwit in een bepaalde stabiele vorm een goede immuunreactie kon opwekken in relevante diermodellen en konden kansrijke kandidaat-vaccins gebaseerd op dit eiwit sneller worden ontworpen, getest en geselecteerd.
Ten vierde werden de krachten vroeg gebundeld. Wetenschappers bij kleine biotechbedrijven (zoals BioNTech) en universiteiten (zoals Universiteit Oxford), die al werkten aan innovatieve vaccinplatforms of expressie en stabilisatie van virale eiwitten, gingen allianties aan met grote farmaceutische bedrijven (zoals Pfizer en AstraZeneca), die veel ervaring hadden met het op de markt brengen en op grote schaal produceren en distribueren van vaccins.
Ten vijfde werd veel tijdwinst geboekt door het in elkaar schuiven van de drie klinische testfasen, de zogenoemde fase I-, II- en III-studies bij vrijwilligers (figuur A en B). Hierbij is het belangrijk om aan te geven dat er in het hele vaccinontwikkeltraject geen stappen overgeslagen werden. Tijdens de eerste klinische testfase I werd in een tiental gezonde volwassen vrijwilligers getest of het vaccin veilig was en of het een immuunrespons opwekte. In fase II werd de optimale dosering in een honderdtal personen uit de doelgroep bepaald en in fase III werd de werkzaamheid, de bescherming tegen COVID-19, en de veiligheid van het vaccin in grote aantallen (10.000+) deelnemers getest. Klinische studies volgden elkaar zo snel mogelijk op, werden gecombineerd of deels tegelijkertijd uitgevoerd in plaats van na elkaar, zoals fase I/II- en/of II/III-studies (figuur A en B). Voorwaarde was wel dat dit niet ten koste ging van de veiligheid.
Ten zesde werd veel tijd gewonnen door te starten met grootschalige vaccinproductie, voordat de resultaten uit de klinische fase III-studies bekend waren. Daardoor konden de eerste vaccins snel geleverd en ingezet worden, zodra de goedkeuring voor toelating tot de markt er was. Dit was mogelijk omdat de (European Commission) en nationale overheden vaccins aankochten tijdens de ontwikkelfase en zo de financiële risico’s voor de fabrikanten afkochten voor het geval dat een vaccin niet werd goedgekeurd. Om de EC en de (Europese Unie)-lidstaten te ondersteunen in hun beslissing over welke vaccins in te zetten, werden wetenschappelijke experts ingezet die tijdens de ontwikkeltrajecten toegang kregen tot vertrouwelijke informatie over veiligheid en immunogeniciteit, als maat voor de werkzaamheid van kansrijke kandidaat-vaccins. Hierbij werd ook gelet op hoe kansrijk het opschalen van het productieproces was.
Tot slot: een laatste stap in een traject van vaccinontwikkeling is het aanvragen van een handelsvergunning bij de registratieautoriteiten, in Europa de (European Medicines Agency). Ook hier werd prioriteit gegeven aan COVID-19-vaccindossiers en het in elkaar schuiven van het proces. Normaal gesproken wordt een dossier van een kandidaat-vaccin pas in behandeling genomen als het hele dossier compleet is. Bij hoge uitzondering kan er gebruik gemaakt worden van een versnelde zogenoemde rolling reviewprocedure, waarbij de dossieronderdelen ingediend worden zodra ze tijdens de uitvoering van de fase III-studie beschikbaar komen. Er kan dus al eerder met het beoordelen van onderdelen van het dossier gestart worden (figuur A en B). Er was in een vroeg stadium dialoog tussen medewerkers van de EMA en de vaccinfabrikanten om verwachtingen op elkaar af te stemmen. Rolling review werd de gangbare procedure voor de COVID-19-vaccins. Om het vele werk te verrichten werd extra personeel ingezet en werd hulp ingeroepen van nationale beoordelaars van medicijnen en vaccins. De totale beoordelingstijd van de EMA voor Comirnaty was 77 dagen, terwijl dit gemiddeld 210 dagen is: een tijdwinst van 4 maanden. Ook hier is het belangrijk te noemen dat ondanks het versnelde ontwikkeltraject, de COVID-19-vaccins aan dezelfde wettelijke vereisten voor productkwaliteit, werkzaamheid en veiligheid, en een positieve balans hiertussen, moesten voldoen, zoals die algemeen gelden voor geneesmiddelen en specifiek voorgeschreven in het TPP voor COVID-19-vaccins.

Figuur A Traditionele vaccinontwikkeling
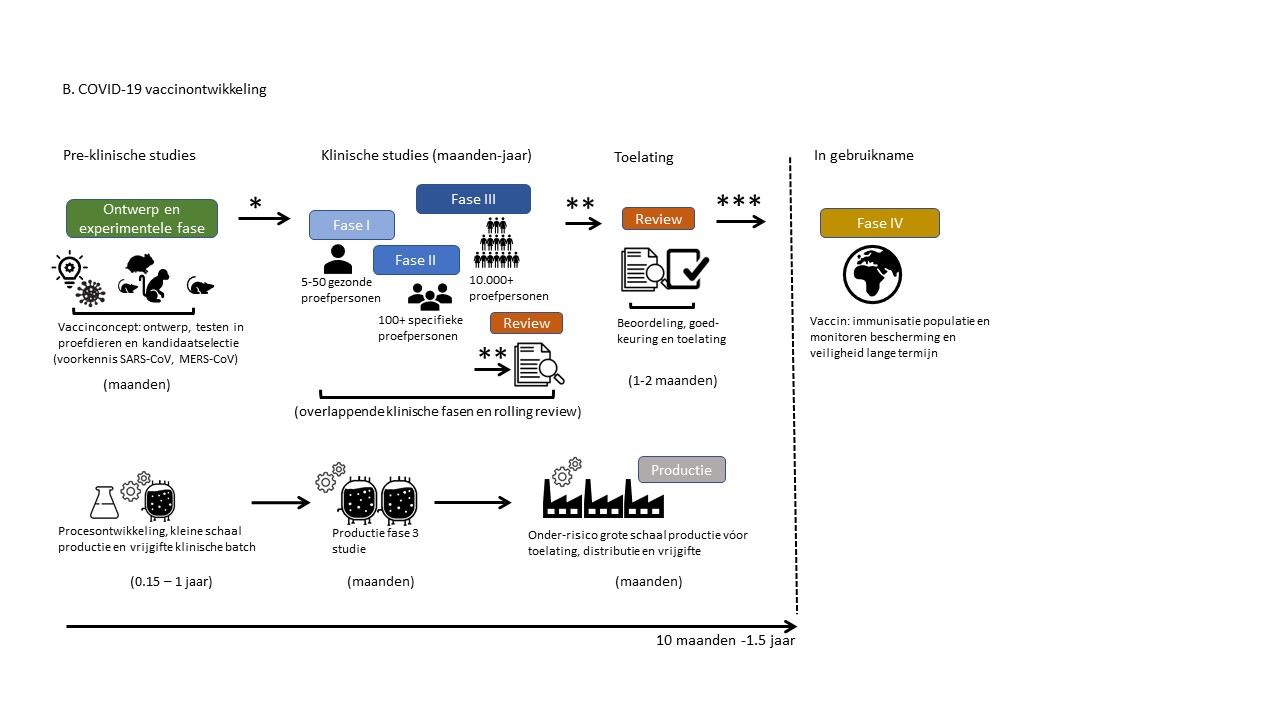
Figuur B COVID-19-vaccinontwikkeling
Zijn er lessen te trekken uit de COVID-19-vaccinontwikkeling die bijdragen aan pandemische paraatheid?
Ja. Draaiboeken voor een volgende pandemie kunnen wat betreft snelle en zorgvuldige vaccinontwikkeling voortbouwen op de volgende conclusies:
- Het maximaliseren van de capaciteit en de samenwerking tussen wetenschappers, fabrikanten, overheden en gezondheidsorganisaties is noodzakelijk.
- Internationale coördinatie door de WHO voor het samenbrengen van wetenschappelijke experts en het vaststellen van eisen aan product- en klinische protocollen is doeltreffend.
- Het snel uitwisselen van wetenschappelijke kennis over de ziekmaker en vaccinplatforms, en het vormen van allianties van wetenschappers en vaccinproducenten leidt tot vroege design en selectie van kansrijke vaccinkandidaten. Sinds COVID-19 weten we dat mRNA in kleine vetbolletjes een over het algemeen veilig en effectief vaccinplatform is met een kort ontwikkeltraject.
- Het in elkaar schuiven van de klinische testfasen I tot en met III en eerder starten met het beoordelen van het registratiedossier leidt tot forse tijdwinst in het ontwikkeltraject.
- Financiële steun door de EC, overheden en publiek-privaat-filantropische samenwerkingen stimuleren wetenschappelijke vaccinontwikkeling en dringen risico’s van voortijdige opschaling van productie door farmacie terug.
- Continue nauwkeurig monitoren van de ziektelast en veranderende eigenschappen van de ziekteverwekker na inzet vaccins op wereldschaal is essentieel om zicht te krijgen op de werkzaamheid van het vaccin op lange termijn en de zeldzame bijwerkingen van nieuwe vaccins.
De COVID-19-pandemie heeft laten zien dat het mogelijk is om binnen een jaar, snel en zorgvuldig een vaccin beschikbaar te hebben. Of dit ook tijdens een volgende pandemie mogelijk is, is afhankelijk van de ziekteverwekker, en of er diermodellen beschikbaar zijn. Of er effectieve vaccins ontwikkeld kunnen worden is mede afhankelijk van overdracht en pathogenese van de ziekmaker.
Infectieziekten Bulletin - december 2023
- Voorwoord VWS bij themanummer Pandemische Paraatheid
- UNITED4Surveillance, samen beter voorbereid op toekomstige gezondheidsbedreigingen in Europa
- Geen pandemische paraatheid zonder persoonsgerichte preventie
- Vaccinontwikkeling tijdens een pandemie: snel en toch zorgvuldig
- De Landelijke Functie Opschaling Infectieziektebestrijding (LFI)
- De basis van de IZB op sterkte met het programma VIP
- Over het Infectieziekten Bulletin